4 — Advanced Pangenome Size Analysis
Biological question: What is the exact size of the pangenome — in number of nodes and in base pairs — specific to each rice subspecies (indica and japonica)?
This use case presents an advanced workflow that combines GraTools commands with the BAM
intermediate files generated during import, using the pysam Python API.
This approach requires familiarity with the command line and Python, but delivers results
much faster than comparable approaches using other PVG tools.
Note
This analysis was completed in less than one hour on the full Asian Rice PVG [Marthe et al., 2025], without any code optimization — considerably faster than the original analysis from [Zhou et al., 2020].
Warning
This use case is intended for power users with bioinformatics expertise. For simpler group-level comparisons, see 3 — Core/Dispensable & Groups.
Workflow Overview
The goal is to identify, for each subspecies, nodes present in at least one sample of that subspecies but absent from all samples of the other subspecies. Their total size in base pairs is then computed using the GraTools BAM file.
Step 1: Prepare Input Lists
Os117425RS1
Os125619RS1
Os125827RS1
Os127518RS1
Os127564RS1
Os127652RS1
Os127742RS1
OsIR64RS1
AzucenaRS1
IRGSP
Os128077RS1
Os132278RS1
Os132424RS1
Step 2: Run specific_groups_sample per Individual
For each indica sample, run a separate command to find nodes present in that sample but absent from all japonica. Create a single-sample file per iteration:
OsIR64RS1
# Repeat for each of the 8 indica samples
gratools specific_groups_sample \
--gfa NewRiceGraph_MGC.gfa.gz \
--samples-list-A single_sample.txt \
--samples-list-B samples_japonica.txt \
--output-csv --suffix OsIR64RS1
Repeat for each of the 5 japonica samples (swapping --samples-list-A and
--samples-list-B).
Step 3: Merge per-Sample Node Lists
After all specific_groups_sample commands, merge the output CSV files to obtain the
full set of nodes exclusive to each subspecies:
# Merge all indica-specific node lists (union)
cat *_specific_Os*RS1.csv | sort | uniq > indica_specific_nodes.txt
# Merge all japonica-specific node lists (union)
cat *_specific_Azucena*.csv *_specific_IRGSP*.csv ... | sort | uniq > japonica_specific_nodes.txt
Step 4: Compute Total Sizes with pysam
GraTools stores node information (including sequence length) in a BAM file generated during
the import step. Use the pysam Python API to sum sizes:
import pysam
bam = pysam.AlignmentFile("NewRiceGraph_MGC.bam", "rb")
with open("indica_specific_nodes.txt") as f:
indica_nodes = set(line.strip().split(",")[0] for line in f if not line.startswith("#"))
total_bp = 0
for read in bam.fetch():
if read.query_name in indica_nodes:
total_bp += read.query_length
print(f"Total indica-specific sequence: {total_bp} bp")
bam.close()
Repeat for the japonica-specific node list and for the shared (core) nodes.
Step 5: Review Results
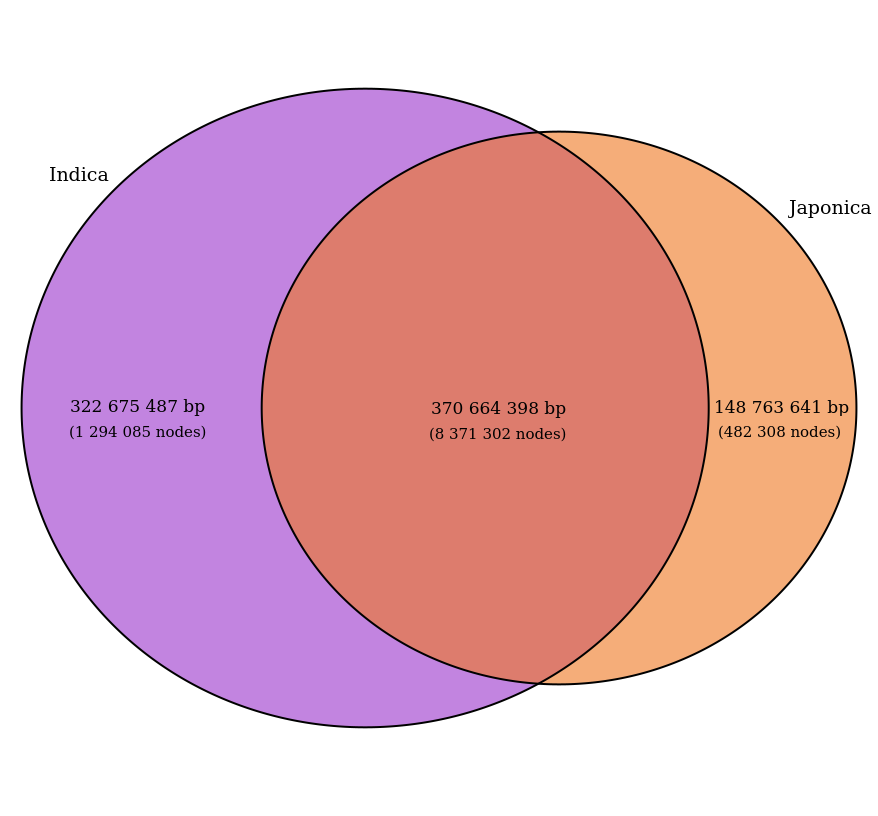
Venn diagram of indica (purple) and japonica (orange) subspecies-specific nodes in the Asian Rice PVG, showing the total sequence size (in bp) of each compartment.
Key findings (Asian Rice, 13 accessions):
The core (shared) portion represents roughly 90–95% of each individual genome, consistent with the expected genome size of 380–410 Mb for Asian rice.
Mean node size in the core: 44 bp; in indica-specific: 249 bp; in japonica-specific: 308 bp.
Most inter-subspecies differences are structural variants, not SNPs [Qin et al., 2021].
The indica compartment is larger, reflecting more indica samples (8 vs. 5).
Summary
Command / Tool |
Description |
|---|---|
|
Find nodes in one sample absent from an entire group |
|
Merge per-sample node lists into group-level node sets |
|
Sum node sizes in base pairs |
See also
3 — Core/Dispensable & Groups — Simpler group comparison with
specific_groups_sampleandpan_ratio1 — Graph Description — Basic graph description
2 — Subgraph & FASTA Extraction — Subgraph and FASTA extraction