gratools pan_ratio
Analyze the ratio between core (shared) and dispensable (specific) segments in your pangenome.
Segments shared by a high percentage or number of samples. This represents the stable, conserved part of the pangenome.
Segments specific to a few samples or unique individuals. This represents the accessory or flexible part of the pangenome.
Options
🛠️ View Command Line Options
$ gratools pan_ratio
Welcome to GraTools version: '1.1.0'
@author: GraTools team's
____ __________ ____
6MMMMMb/ MMMMMMMMMM `MM
8P YM / MM \ MM
6M Y ___ __ ___ MM _____ _____ MM ____
MM `MM 6MM 6MMMMb MM 6MMMMMb 6MMMMMb MM 6MMMMb\
MM MM69 " 8M' `Mb MM 6M' `Mb 6M' `Mb MM MM' `
MM ___ MM' ,oMM MM MM MM MM MM MM YM.
MM `M' MM ,6MM9'MM MM MM MM MM MM MM YMMMMb
YM M MM MM' MM MM MM MM MM MM MM `Mb
8b d9 MM MM. ,MM MM YM. ,M9 YM. ,M9 MM L ,MM
YMMMMM9 _MM_ `YMMM9'Yb_MM_ YMMMMM9 YMMMMM9 _MM_MYMMMM9
\ / /
/''A''\ /''''''\ / /''''A'''''\
...GC| |..ATG...C...CG...T....TAG..'..GC.| |...
\..C../ \.............../ \...TATA.../
Please cite our gitlab: https://forge.ird.fr/diade/gratools.git\
Usage: gratools pan_ratio [OPTIONS]
Aliases: ratio
This command analyzes segments in the GFA to determine the ratio of core
segments (shared by almost all samples) versus the dispensable ones (present
in a smaller subset of samples). The thresholds for defining 'core' and
'dispensable' can be specified as an absolute number of samples or as a
percentage of the total number of samples embedded in the GFA. A filter on
segment length can also be applied. Results are displayed in the terminal and
saved to a CSV file. This command relies on a pre-existing GraTools import of
the input GFA.
For more details, see the full documentation:
https://gratools.readthedocs.io/en/latest/commands/pan_ratio.html
Pan Ratio Ratio Options:
-g, --gfa PATH
Path to the input GFA file (e.g., myGraph.gfa or myGraph.gfa.gz).
[required]
-o, --outdir DIRECTORY
Output directory for GraTools results. If not specified, results are
typically placed in a subdirectory within the GFA file's parent directory
(e.g., 'GraTools-output_<gfa_name>').
-su, --suffix TEXT
Custom suffix to append to output filenames. If not provided, a default
suffix will be generated based on the command line parameters.
--input-as-number / --input-as-percentage
Specify whether --shared-min and --specific-max are absolute numbers or
percentages. [required]
-sm, --shared-min TEXT
Minimal number/percentage of samples embedded in for a segment to be
'core'. [required]
-spm, --specific-max TEXT
Maximal number/percentage of samples embedded in for a segment to be
'dispensable'. [required]
-fl, --filter-len INTEGER
Minimal segment length (bp) to be included in the analysis. A value of 0
means no length filter. [default: 0]
Logging Options:
-vv, --verbosity [DEBUG|INFO|ERROR]
Set the logging verbosity level. [default: INFO]
-l, --log-path DIRECTORY
Directory where the log files will be saved. If not specified, logs will be
placed in the main output directory (or in a default GraTools log
location).
Performance Options:
-t, --threads INTEGER
Number of threads to be used for parallelizable operations. [default: 1]
Other options:
-h, --help
Show this message and exit.
Usage Example
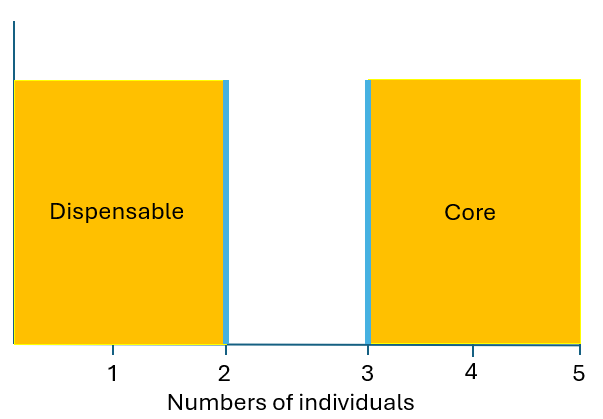
In this example, we analyze a GFA with 5 samples.
Core threshold: Shared by at least 4 samples (
--shared-min 4).Dispensable threshold: Present in 2 or fewer samples (
--specific-max 2).Filter: Only segments ≥ 50 bp are considered for the filtered stats (
--filter-len 50).
$ gratools pan_ratio -g Og_cactus.gfa.gz --input-as-number \
--shared-min 4 --specific-max 2 --filter-len 50 --threads 4
Output Summary:
─────────────────────────────────────────── Summary ──────────────────────────────────────
Total segments in GFA: 2,354,995
Total segments analyzed: 2,354,995
Total segments passing length filter (≥ 50bp): 210,046 (8.92%)
Core vs. Dispensable Segments — Og_cactus
╭────────────────────────────────────────────────────┬───────────┬───────────┬────────────╮
│ Category │ Count │ Total │ Percentage │
├────────────────────────────────────────────────────┼───────────┼───────────┼────────────┤
│ Shared (Core) - Raw │ 1,162,062 │ 2,354,995 │ 49.34% │
│ Specific (Dispensable) - Raw │ 891,345 │ 2,354,995 │ 37.85% │
│ Shared (Core) - Filtered (Length >= 50bp) │ 195,840 │ 210,046 │ 93.24% │
│ Specific (Dispensable) - Filtered (Length >= 50bp) │ 8,602 │ 210,046 │ 4.10% │
│ Segments Filtered Out by Length │ 2,144,949 │ 2,354,995 │ 91.08% │
╰────────────────────────────────────────────────────┴───────────┴───────────┴────────────╯
Understanding the Results
Percentage based on all segments in the GFA.
Shared (Core) - Raw: The number of core segments as a percentage of all segments in the GFA.
Specific (Dispensable) - Raw: The number of dispensable segments as a percentage of all segments in the GFA.
Percentage based only on filtered segments (≥ --filter-len).
Shared (Core) - Filtered: The number of core segments that also meet the length filter, as a percentage of only the filtered segments. This shows the composition of the longer segments.
Specific (Dispensable) - Filtered: The number of dispensable segments that meet the length filter, as a percentage of only the filtered segments.
Filtered Out: The total number and percentage of segments that were excluded from the “Filtered” analysis because they were shorter than the –filter-len value.
Process & Logic
The --shared-min (-sm) and --specific-max (-spm) options specify the number of samples a segment must be found in to be considered part of the “core” or “dispensable” genome, respectively.
Input mode: The user must specify whether the thresholds are :
Use --input-as-number.
Example: --shared-min 4 means the segment must be in 4+ samples to be Core.
Use --input-as-percentage.
Example: --shared-min 90% means the segment must be in 90% of samples.
Example: For the command gratools pan_ratio –input-as-number –specific-max 2 –shared-min 4, the minimum number of samples for a segment to be “core” is 4, and the maximum number of samples for it to be “dispensable” is 2.
The --filter-len (-fl) option excludes short, often uninformative segments from the “Filtered” analysis. This helps focus on structural variations and significant genomic regions.
In many pangenome graphs, a large number of segments are very short (1-50 bp). Use --filter-len to see the “real” structural core genome, as short segments can sometimes skew the percentage towards core due to graph complexity.
📑 Quick Links
Command Import: gratools import
Related Tool: gratools depth_nodes_stat